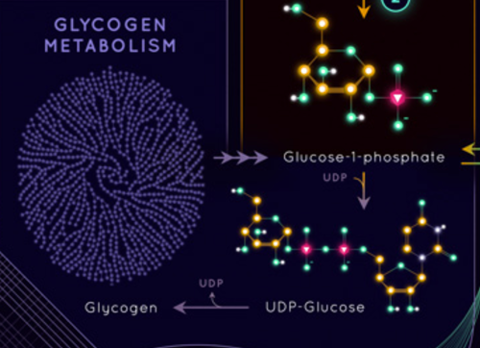
Just look at that drawing of Glycogen … that’s purty.
As the George Henderson mentioned in the comments it misses the primary entry point for even chained fats.
#Fats and Ketones
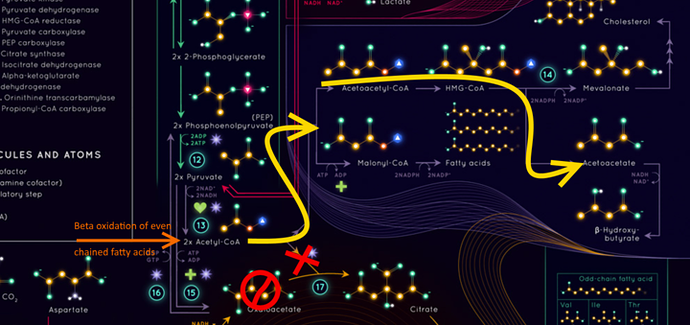
Someone asked me the other day how ketones are made and it looks a little like this ( I had to mark up the infographic a bit … I’m no artist so apologies for the graffiti.);
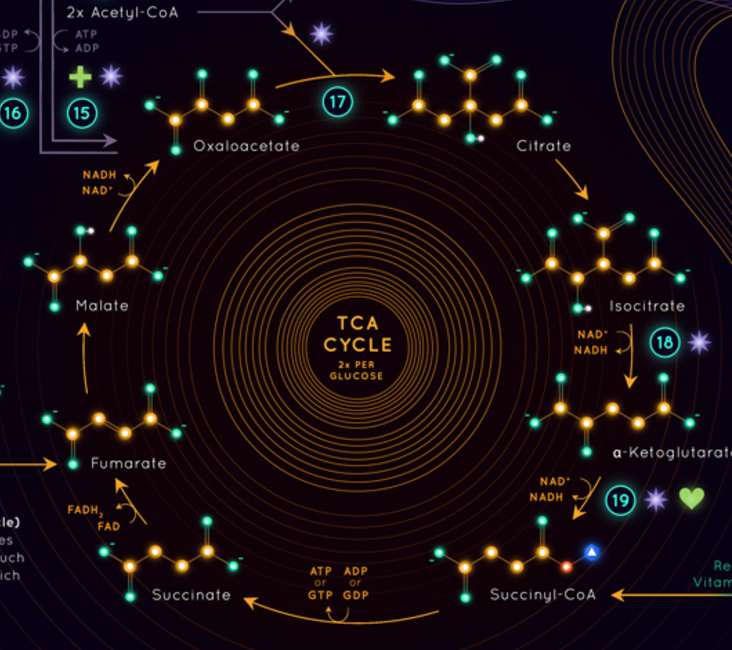
It’s worth looking at how fats get into the system in a process called beta oxidation. Even chained fatty acids are chomped up into 2 carbon unit Acetyl molecules which are bonded to a carrier called Coenzyme A … or CoA, to make Acetyl-CoA. At this point it enters the TCA (or kreb) cycle which is a series of 9 reactions (although only 8 are shown in this infographic) that makes carbon dioxide and energy (12 ATPs for every Acetyl-CoA).
This process adds our 2 carbon acetyl to a 4 carbon Oxaloacetate to make a 6 carbon Citrate and then goes through this series of steps to become a 5 carbon alpha-ketoglutarate and eventually becoming a 4 carbon Oxaloacetate again - ready for the next 2 carbon acetyl.
BTW old mate Glucose ALSO gets into this cycle at the same point, it’s cleaved into 2 pyruvates a 3 carbon molecule which is split into an acetyl and a CO2 -> and then that acetyl goes through the same process. This extra release of CO2 when burning glucose is why the respiratory quotient (CO2 breathed out / O2 breathed in) is higher (1.0) for glucose burners, than for fat burners (0.7).
The Kreb cycle is how we make the majority of our ATP we use for energy, it’s also where we make the rest of our CO2, but not precisely where we use our Oxygen (that happens in the Electron transport chain).
So you know that we MAKE glucose when we don’t eat it - Gluconeogenesis? That process steals one of those Kreb cycle metabolytes called Oxaloacetate. What that does is it throws a spanner in the cycle (the ), and it does it right before the Acetyl-CoA goes into the cycle ( the ). Those Acetyls coming from the liver trying to burn fat then follow the yellow arrow
Those Acetyls are shunted off on a side route following the Yellow arrows to eventually make the first ketone Aceto-Acetate. This is happening in our liver cos that is where Glucogenesis happens, the Oxaloacetate stolen, the AcetoAcetate made. At this point it spills into circulation to eventually be taken up by muscle cells that convert it into beta-hydroxybutyrate and then it can go to our brains to make them less anxious about the dire lack of glucose.
Fatty acids aren’t exactly chomped into acetyls and then CoA added.
What happens is the first one is added outside the Mitochondria while it gets shuttled into the matrix, and then eventually that long chain has it’s terminal Acetyl-CoA split off and a new CoA added to the end of the chain remaining.
This video lecture on the subject I found useful, and I wanted somewhere to keep it … why not here.
@richard, this is great stuff! thank you! I love seeing biochemical breakdown explained this way.
I’d like to have a better understanding of what the bottlenecks are in the system. It is good to see the biochemical reactions, showing what ingredients/factors feed into each step, however, I’d like to know which is in complete abundance in the body versus limit-constrained in some individuals. By applying a “theory of constraints” (i.e. also known as TOC) approach, like engineers do when analyzing systems, I think we can develop a better strategy to overcome deranged metabolisms.
On this forum, we see people embark on keto/fasting regime and then lose weight in a blink, while others need to stick to it for ages to slowly lose. It leads me to believe that the biochemical reactions presented in the materials you linked above are subjected to bottlenecking factors and perturbing the outcome of the metabolic process.
For example, in the video you posted, the teacher demonstrates how carnitine participates in the shuttling of the ex-fatty acid complexed molecule through the mitochondria wall. If one considers this as ONE process step, in a sequence of MANY process steps, then the hypothesis is that this process step may have some bottlenecking factors at play. For example…what happens if carnitine is insufficient supply? Or…what if there is insufficient healthy mitochondria? What other factors on the mitochondria wall discourage passage of carnitine-complexed molecules? What other physical or biochemical barriers impede the transport of carnitine-complexed molecules into the mitochondria? I’d like to see a diagram that shows every possible bottlenecking factor suspect at every process step.
I love seeing these step-by-step explanations of the process steps of breakdown of fats into energy.
However, I really really really wished there was a step-by-step explanation of “suspect bottlenecking factors” at each process step. This way, we can all look at the process steps, and scientifically run n=1 experiments, in order to see if we can isolate the bottlenecked process steps in our bodies. This can then be followed by the subsequent n=1 experiments to devise a de-bottlenecking tactic for that bottlenecked process step.
I realize I’m thinking like an engineer…breaking down the elephant into bite size pieces for root cause analysis. This is what engineers do. When there is a failure in a system, they break the system down into pieces, and logically analyze each piece where things can go wrong. By eliminating factors of what may have gone wrong, they end up with a very small group of suspects, and then they isolate each variable and test the heck out of it to prove “who screwed up the system” and what exact conditions existed for that suspect variable to screw up the system. But, I think this way of doing root cause analysis does not exist in the medical or biochemistry fields. I see biochemistry diagrams bundled together into a complicated molecular “stew”. and then with the statement…“we are all unique snowflakes”, or "our hormones change things on the fly, and “we cannot control the mechanisms because everything changes anyway”. I call bullshit on that. I have a hunch that breaking down these biochemical process steps, identifying the bottlenecking factors, and then targeting the factors with de-bottlenecking tactics, will lead us to a more efficient effort of undoing the metabolic damage. This would be a much better way than randomly trying a bunch of tactics (avoiding dairy, changing macro nutrients, IF tactics, EF tactics, changing salt content in food, skipping breakfast, exercising, more protein, less protein, grass-fed versus non-grass fed, blah blah blah blah blah etc). Even though this keto community provides a basic framework as a “starter’s template” to embark on the keto journey…there is a lot of random n=1 going on, with the hope something works for all the “unique snowflakes”. I’d like to see a more engineer’s theory of constraints and root cause analysis approach. And stop relying on “experts” telling us we are “unique snowflakes”.
Fiorella steps off the soap box and quietly walks away…
I looked into some of the rate limiting steps involved in Gluconeogenesis for the “Steak therefore Cake” post;
I don’t think anyone has taken a theory of constraints approach to energy biochemistry as a system. You might be right there may be a fertile territory there.
Those are all intriguing questions.
I’m going to try answer some (and a few you didn’t ask) using my intuited synthesis (with a few references for the bits I can prove)
So I understand that fatty acids with 10 or fewer carbons will diffuse straight across the mitochondrial “cell” wall … so short and medium fatty acids are not affected by any inhibition in the carnitine shuttle.
When the shuttle is bottle necked then there are backup organelles called peroxisomes that cleave 12 carbon and larger fatty acids into shorter chained ones … but this process generates heat and hydrogen peroxide. This may be a very old holdover from before our ancestor captured and enslaved the mitochondria. We may have retained it to maintain flexibility in situations when mitochondria went haywire.
We can store some fatty acids in lipid droplets in cells and they may mechanically inhibit the insulin mediated glucose receptor (GLUT4) from getting to the cell wall - so lipid backup in the cell produces insulin resistance … which makes sense from a systemic standpoint.
The other control point that is interesting from a systemic standpoint is … what switches us between using fat and using glucose (and making fat with any we don’t use) is the presence of Malonyl-CoA and it’s mechanism is to inhibit the carnitine shuttle shutting off the spigot of fat entering the mitochondria so it can focus on other sources of energy.
It turns out that this signal shutting off the freeway for long chained fats into our mitochondria is insulin going high. Well … the above paper doesn’t take it to that conclusion, they just say glucagon is the on signal for ketogenesis. The approximate sequence is Insulin goes high, glucagon goes low, via AMPK that unblocks synthesis of Malonyl-CoA - that turns off the carnitine shuttle stopping fats from entering in bulk to be burned. When insulin goes low, glucagon rises, via AMPK blocks the synthesis of malonyl-CoA … and we make ketones instead.
The TL;DR version if when Insulin is high we have to get energy from burning glucose and protein and medium chained fats - and we make long chained fats for storage. When Insulin is low the tollgate raises on the long chain fat freeway and we switch to burning fats at full speed.
This explains the paradox of the insulin resistant person who is lethargic, hungry and apparently has plenty of stored energy. They aren’t able to use fats efficiently with that much insulin in circulation.
This also explains why we make fat in every cell when insulin is high - because our cells are either making ketones or fat (denovo) from fuel in excess to the capacity of our krebs cycle. And if our fat cells are healthy, at a signal from insulin they hoover it all up for storage.
The other thing I believe this explains is why very deranged people lose a lot of weight on keto then stall. In the starting glucose burning state our fat cells have become insulin resistant and unable to hold their fat, spilling it over into circulation. What is actually stopping us using the fat for energy is high insulin is inhibiting the carnitine shuttle in all our cells.
Keto usually lowers insulin sufficient to enable use of fat for energy and we’re off to the races … until out fat cells get rid of enough of their cargo to become insulin sensitive again. If we can lower our insulin below a certain point there is not plateau … we just keep cruising lower till we hit other constraints on fat loss. If we cant lower insulin below the point needed to release the inhibition on our now healthy fat cells … plateau city. And then the goal is to find ways to get insulin below that 13 mIU/l point above which fat cells are inhibited from releasing energy for use.
Carnitine is also interesting in that it has been marketed as a “fat burning” supplement for quite some time, which may indeed be true if you are deficient. People who avoid carnitine-rich foods might fit into this category for example. I suspect the more carnivorous among us get a more abundant supply from red meat etc. but it doesn’t do us any good if insulin breaks the mechanism.
I’ve always wondered what would happen if we made an electronic or code model of metabolic pathways, then tweak or break parts of it to see systemic consequences?
If engineers can today build a simulator for pilots to train how to fly a plane under competing conditions (weather, flying obstacles, different terrain, etc) then why doesn’t a simulator exist for human metabobilism? It makes no sense to me. The technology exists where engineers are at work creating innovations, and a vacuum of knowledge exists where engineers don’t traditionally “play”, like medical industry.
It’s like if medical industry ran aviation industry, a pilot who runs into a difficult situation in the air would have to call an “expert” for advice to get out of a dangerous storm, or read a stack of peer reviewed papers to figure out which best move to make to rescue the flight. The information in medical industry is so fragmented and disorganized that it takes way too long to make optimal decisions at the speed a pilot simulator could make. The info exists. It’s just horribly disorganized. And the excuse “it’s complicated” is used to cover up the disorganization.
I am sooooooooo looking forward to artificial intelligence entering the medical industry space. It will be a game changing technology that will forcibly organize the data into simulator capability.
This is another of the many great and fascinating threads on ketogenicforums.com. There is so much to learn about our metabolism, and a fairly large number of things to become familiar with and internallize, just to have many of the basics make sense to us (if that matters).
Richard, thank you so much for the time and effort you put into this stuff.
Richard, you do fabulous work my friend. This is an excellent description of Oxidation of fatty acids. Thank you for sharing. I have also used this one to drill the biochemistry into my thick skull. https://youtu.be/gQYCCiGe9bo
I found a quote in a paper that describes the carnitine limitation due to insulin, without all the molecules:
In the adult mammal, ketogenesis and the utilization of KB are regulated at several key steps that prevent substantial amounts of KB from being produced under normal feeding conditions (FIG. 2). Ketogenesis requires abundant circulating free fatty acid (FFA) levels for oxidation in the liver. Thus, conditions such as elevated insulin signaling that limit adipocyte release of FFA, hamper KB production. Once in the hepatocyte, FFA may undergo either oxidation in the mitochondria or re-esterification to triacylglycerols or phospholipids. Carnitine palmitoyltransferase-I (CPT-1) functions to transfer activated FFA into the mitochondria and is a major regulator of the choice between oxidation and esterification. In the nourished state, the presence of insulin signaling inhibits CPT-I, and few KB are produced. In addition, KB may regulate FFA release as part of a feedback loop. -hydroxybutyrate (BHB) has been demonstrated to activate the receptor HM74a leading to reduced FFA secretion from adipocytes. HM74a is better known as the niacin receptor, a potent lipid-lowering agent. http://www.sciencedirect.com/science/article/pii/S1933721308000937
So, ketone production is limited by insulin-FFA, insulin-CPT1 and BHB-FFA.


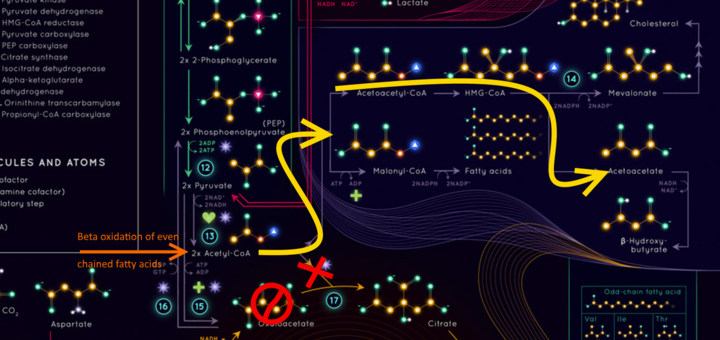
 ), and it does it right before the Acetyl-CoA goes into the cycle ( the
), and it does it right before the Acetyl-CoA goes into the cycle ( the  ). Those Acetyls coming from the liver trying to burn fat then follow the yellow arrow
). Those Acetyls coming from the liver trying to burn fat then follow the yellow arrow