Disclaimer: I do not have a medical or biology background, so feel free to point out any errors or misunderstandings. However, please reference publications for refuting arguments, not opinions.
In June of 2021 I was told that my A1C was 10.3 and my doctor wanted to increase my metformin and other diabetic medications. I did some research on diabetes (again, as I had previously tried without much success) and got quite scared upon reading the possible side effects of diabetes with such high blood glucose levels. As with most, before I started eating a ketogenic diet I researched. I wanted to understand if a ketogenic diet was a safe way of eating, and if it could reverse my type II diabetes (T2D). As with most research, you find a few videos/articles which (hopefully you understand them) then lead to other papers/articles/videos and your knowledge and understanding increase as you watch/read/listen to the available information.
My initial research led me to the conclusion (based on listening to Ben Bikman in particular) that people with type II diabetes could have higher than normal blood glucose from protein intake while on a very low carb diet. In hindsight it appears Ben is referring not to an actual sharp rise in glucagon (and then blood glucose) due to hyper sensitive glucagon reaction to certain amino acids https://www.youtube.com/watch?v=MEzAvos1jak&t=111s [although he also notes that T2D may have more GNG due to hyperglucagonemia from protein]. When I mentioned this on ketogenic forums, I was told that I was mistaken and that protein did not increase blood sugars and that diabetes had zero effect on this process. I was further confused when the vast majority of posters “liked” or agreed with the statement that protein did not influence blood sugars in general and also apparently specifically for people with T2D as well. As I love meat, I happily started to eat a lot of meat/protein with the thought that my blood glucose would not go up (as per this forum). I then measured and my blood sugars had gone up noticeably. Subsequently I spent some time “proving” to myself that my blood sugars were indeed directly linked to my protein intake CGM data, Carnivore and Blood Glucose levels versus protein intake . The reality is that if your measurements do not coincide with the theory, then either the theory is incorrect or the measurements were not taken properly. I repeated the experiment a third time and got the same results. I needed to find out why the theory was not matching the experiment (on these boards since it has since become clear in the literature that protein does indeed affect blood glucose levels in T2D in particular - but also potentially in general as well).
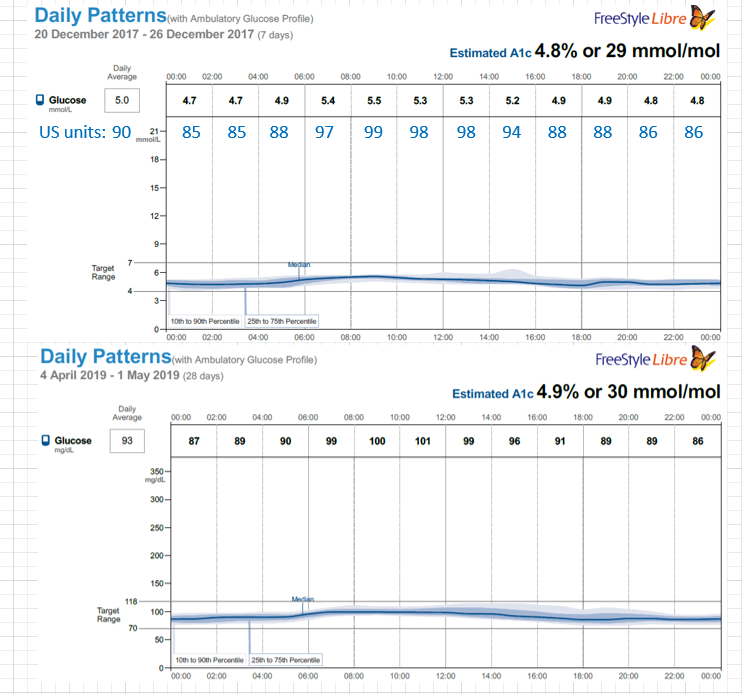
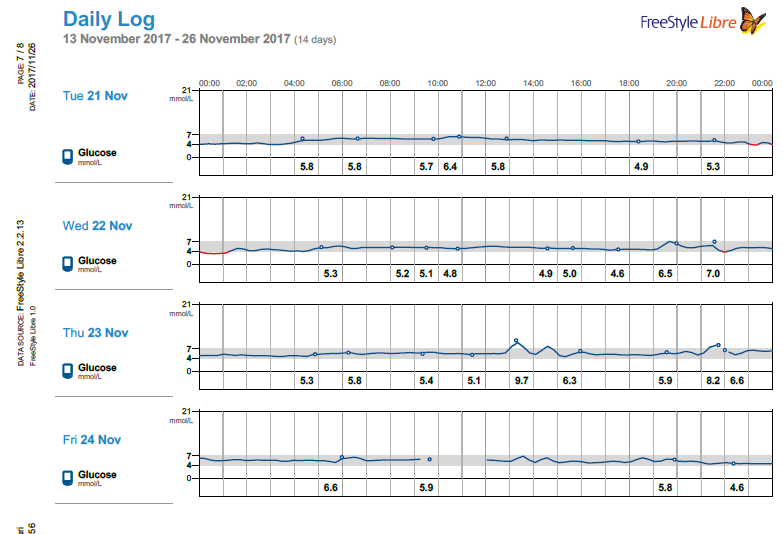
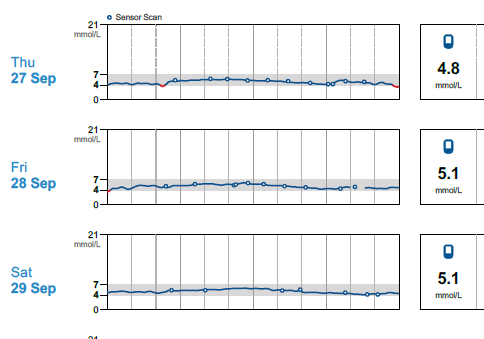
Months later I read a paper on gluconeogenesis, and again, understood the results a bit differently that others who read the same paper. This lead me to re-investigate the topic, which then lead me to a plethora of research papers on the topic of both gluconeogenesis and blood glucose from protein, which I had somehow missed during my initial searches months earlier. Posts within these boards discussed increased blood glucose from protein for people on a ketogenic diet: WTF Keto! and Gluconeogenesis: I keep hearing that it's only demand driven, can that be right? and there was a blog as well from Richard https://blog.2keto.com/steak-cake/ with a reference to https://www.ketogains.com/2016/04/gluconeogenesis-wont-kick-you-out-ketosis/. A quote from Richard’s blog “Anyway this explains how Adam and Jimmy can observe that people who eat protein see a drop in ketones and a significant increase in glucose – and yet GNG is still not a substrate limited process.” I wish I had found these posts and blogs earlier just to fast track some of my learning. While blood sugar rising due to protein ingestion is not new, this is not exactly what I measured in my testing. Yes, my blood sugar may rise slightly shortly after eating, but it was 12 hours later (after a sleep), that my FBS would rise in the morning and my BS levels were consistently higher the following day. As noted in my experiment, my daily average for blood glucose (using a CGM) was 5.2 mmol/L (93.6 mg/dL) when eating 1.5 g of protein/ kg bodyweight the previous day and elevated to 6.0 mmol/L (108 mg/dL) at 4.25 g protein/kg bodyweight in a linear manner. An increase in the total daily glucose in the blood of 19.2 mmol/L (345.6 mg/dL) over the day (if looking at area under the curve). While some people would argue this is not a “spike” since it is a general rise in the baseline, I consider that increase to be a significant daily spike (since I am sure if it happened over a few hours, people would call it an extremely large spike). Excessive protein ingestion has to be absorbed and then utilized or disposed [4].
In order to understand the reasoning behind this, the glucagonogenic viewpoint of GNG as opposed to the insulinogenic viewpoint may be useful here [2]. The concept that blood sugar needs to be low with low insulin to create a demand for gluconeogenesis is only looking at a small part of the issue. GNG is an extremely complex process which depends on many facets [16]. One of the major signals that promotes GNG is glucagon [20]. In the past, glucagon was thought to be dependent on insulin levels and not as an independent hormone with other duties other than counter acting insulin. Six years ago the glucagon and liver-alpha-cell axis was not well documented, but the theory has since been accepted [7, 8, 11, 17, 21, 25, 32]. Some things worth mentioning is that the digestion of proteins increases insulin (up to 6 times) and glucagon (up to 8 times), such that there is at least as great if not greater glucagon response than insulin [24] and that glucagon release is up to seven times more dependent on amino acid levels as compared to glucose levels [32]. Some authors suggest that glucagon releasing alpha cells are in fact primarily amino acid sensors [21]. Using this concept, one could easily understand that the process of eliminating abundant amino acids from the bloodstream is controlled most by glucagon and this has a side effect of increasing GNG as well. It has been accepted that GNG is a very complex process that is thought to be demand driven [16]. I would argue that the demand is driven by supply (protein consumption) and then by definition this is a supply driven model for GNG (demand required due to oversupply). Regardless of your position on supply or demand driven, the feedback loop continues to keep glucagon elevated for more than 5 hours (at least 8) after protein ingestion [1] while insulin returns to base values before 5 hours. The contribution of dietary protein to GNG has only been studied in the context of SAD and therefore the implications of a ketogenic or carnivore diet has not specifically been addressed. In this context however, it was found that protein does not contribute to GNG when ingested with carbohydrates, and for SAD participants, only 8% of protein was converted into glucose [5] without carbohydrate consumption. In addition however, these studies only considered a few hours after ingestion to be of value when considering GNG [24]. Certainly studies up to at least 8 hours would be better, and from my experiments, I would rather see 12 to 16 hours after ingestion. Part of the reason for most studies to conclude after only a few hours, is based on the fact that there is limited protein storage within the body (nothing similar to fat deposits), and therefore the concept of temporary protein storage through skeletal bone/muscle is being disregarded. More studies into GNG from longer term high protein intake should be undertaken.
As I am a type 2 diabetic, it could also be that protein ingestion raises my blood sugar but it would not raise the blood glucose of a highly insulin sensitive individual. In general, someone with type 2 diabetics is thought to have higher blood glucose due to hyperglucagonemia which instigates GNG [6, 10, 13,17, 18, 27, 28, 32]. This is also true however, whether the person has steatosis or insulin resistance, regardless of the presence of elevated glucose [25]. In this way, one could easily find a scenario whereby someone with IR but who is not diagnosed with T2D, has higher blood glucose after protein ingestion. Regardless, it is well accepted that protein intake increase blood glucose levels for people with T2D. While I have no published papers to corroborate my thinking, I highly suspect that an insulin sensitive person eating 4.25 g of protein / kg bodyweight would also have a (slightly) higher FBS than normal the next day, hopefully Ben Bikman or others will test thoroughly in the next few years.
-
Incretin and Islet hormonal responses to fat and protein ingestion in healthy men (2008) https://journals.physiology.org/doi/full/10.1152/ajpendo.90233.2008
-
Glucagonocentric restructuring of diabetes (2012) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3248306/
-
Dietary protein and the blood glucose concentration (2013) https://diabetesjournals.org/diabetes/article/62/5/1371/42864/Dietary-Protein-and-the-Blood-Glucose
-
protein turnover, ureagenesis and gluconeogenesis (2013) https://pubmed.ncbi.nlm.nih.gov/22139560/
-
Dietary Proteins Contribute Little to Glucose Production , Even Under Optimal Gluconeogenic Conditions in Healthy Humans (2013) https://diabetesjournals.org/diabetes/article/62/5/1435/42876/Dietary-Proteins-Contribute-Little-to-Glucose
-
Nutrient Regulation of glucagon secretion: involvement in metabolism and diabetes (2014) https://www.cambridge.org/core/journals/nutrition-research-reviews/article/nutrient-regulation-of-glucagon-secretion-involvement-in-metabolism-and-diabetes/C245597B55CE223395A91EF9FFD6D7BA
-
The biology of glucagon and the consequences of hyperglucagonemia (2016) https://www.futuremedicine.com/doi/full/10.2217/bmm-2016-0090
-
Glucagon’s effect on liver protein metabolism in vivo (2017) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5625084/
-
Insulin and Glucagon – Partners for Life (2017) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6061217/
-
Regulation of hepatic glucose metabolism in health and disease https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5777172/ (2017)
-
Glucagon and Amino Acids are linked in a Mutual Feedback Cycle (2017) https://diabetesjournals.org/diabetes/article/66/2/235/35269/Glucagon-and-Amino-Acids-Are-Linked-in-a-Mutual Glucagon and the liver – alpha – cell axis https://web.archive.org/web/20190430084333id_/http://diabetes.diabetesjournals.org/content/diabetes/66/2/235.full.pdf
-
Rate of Appearance of Amino Acids after a Meal Regulates Insulin and Glucagon Secretion in Patients with Type 2 Diabetes: A randomized clinical trial (2018) https://academic.oup.com/ajcn/article/108/2/279/5049681?login=false
-
Hyperglucagonemia correlates with plasma levels of non BCAAs in patients with liver disease independent of T2 diabetes. (2018) https://pubmed.ncbi.nlm.nih.gov/28971838/ or full at https://journals.physiology.org/doi/full/10.1152/ajpgi.00216.2017?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org
-
Mechanisms of Insulin Action and Insulin Resistance (2018) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6170977/
-
Role of glucagon in protein catabolism (2018) https://pubmed.ncbi.nlm.nih.gov/29877875/
-
Unravelling the Regulation of Hepatic Gluconeogensis (2018) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6353800/ Hepatic gluconeogenesis is a complex and genetically heterogeneous process modulated on multiple levels (1). Although there are growing data on the identification of transcription control of gluconeogenesis, which is a relatively slow process, limited information is available regarding the acute control of gluconeogenesis. Indeed, hepatic gluconeogenesis is suppressed rapidly following the intravenous administration of either galegine or metformin in rat; this is inconsistent with that of transcriptional regulation mechanisms. Regulation of hepatic gluconeogenesis can also be controlled through the regulation of substrate supply and allosteric regulation of enzyme activity. For example, glucagon can stimulate hepatic gluconeogenesis by inhibiting the liver-specific PK and induces the phosphorylation of the fructose-2,6-bisphosphatase 2 (PFK2). Phosphorylated PFK2 functions as phosphatase to decrease the production of fructose-2,6-bisphosphate, resulting in an inhibitory attenuation to the gluconeogenic enzyme FBPase. Additionally, phosphorylated PFK2 suppresses GK by promoting its nuclear translocation. Overall, identifying key regulators that control gluconeogenesis could provide novel ways to treat type 2 diabetes.
-
Evidence of a liver-alpha cell axis in humans: hepatic insulin resistance attenuates relationship between fasting plasma glucagon and glucanotropic amino acids (2018) https://link.springer.com/article/10.1007/s00125-017-4535-5 “Specifically, we found in 1408 individuals with normal and impaired glucose regulation that the fasting plasma levels of four glucagonotropic amino acids were inversely associated with fasting glucagon levels independent of age, sex, BMI and peripheral insulin resistance (ISI0–120), and that the associations were modified by hepatic insulin resistance (HOMA-IR).” “In conclusion, higher fasting plasma glucagon concentrations were associated with lower concentrations of certain non-BCAAs including alanine, tyrosine and glutamine, and with higher concentrations of BCAAs, in a large study population covering a broad glycaemic range from normal glucose tolerance to screen-detected type 2 diabetes. A liver–alpha cell axis may therefore exist in humans by which glucagon controls plasma levels of amino acid through hepatic gluconeogenesis and ureagenesis. Importantly, impaired liver function (increased HOMA-IR) weakened this relationship, resulting in higher amino acid levels and ensuing hyperglucagonaemia. If validated in other studies, a glucagon–alanine index may be a useful surrogate marker of glucagon receptor signalling in humans.”
-
E2F1 Promotes hepatic gluconeogenesis and contributes to hyperglycemia during diabetes (2018) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6001358/ In conclusion, the results presented here, along with our previous finding that E2F1 contributes to hepatic steatosis, demonstrate that this transcription factor critically contributes both to hyperlipidemia and hyperglycemia during insulin resistance. This suggests that an interesting therapeutic window exists to simultaneously target these two key metabolic pathways that are abnormally increased in type 2 diabetes.
-
CPT1a-Dependent Long-Chain Fatty Acid Oxidation Contributes to Maintaining Glucagon Secretion from Pancreatic Islets (2018) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6581793/ Here, we show that glucagon secretion in low glucose conditions is maintained by fatty acid metabolism in both mouse and human islets, and that inhibiting this metabolic pathway profoundly decreases glucagon output by depolarizing α cell membrane potential and decreasing action potential amplitude.
-
Methods and Guidelines for Measurement of Glucagon in Plasma (2019) https://www.researchgate.net/publication/336929835_Methods_and_Guidelines_for_Measurement_of_Glucagon_in_Plasma
-
A primary role for alpha-cells as amino acid sensors (2019) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7085241/ The liver–α-cell axis is conserved across vertebrate species. Interrupted glucagon signaling impairs liver catabolism of amino acids, leading to hyperaminoacidemia. Remarkably, high levels of amino acid stimulate glucagon secretion and under sustained hyperaminoacidemia can promote α-cell hyperplasia. However, many unanswered questions remain surrounding the mechanism of amino acid regulation of α-cell function.
-
Glucagon Receptor Signaling and Lipid Metabolism (2019) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6491692/ Glucagon may, aside from its physiological actions on glucose and amino acid metabolism, also be important for lipid metabolism via effects on hepatic beta-oxidation and lipogenesis, and potentially increased lipolysis in adipocytes.
-
Glucagon Physiology (2019) https://www.ncbi.nlm.nih.gov/books/NBK279127/ Glucagon is a glucoregulatory peptide hormone that counteracts the actions of insulin by stimulating hepatic glucose production and thereby increases blood glucose levels. Additionally, glucagon mediates several non-glucose metabolic effects of importance for maintaining whole-body energy balance in times of limited nutrient supply. These actions include mobilization of energy resources through hepatic lipolysis and ketogenesis; stimulation of hepatic amino acid turnover (and related ureagenesis). Also, glucagon has been shown to increase energy expenditure and inhibit food intake, but whether endogenous glucagon is involved in the regulation of these processes remains uncertain. Glucagon plays an important role in the pathophysiology of diabetes as elevated glucagon levels observed in these patients stimulate hepatic glucose production, thereby contributing to diabetic hyperglycemia.
-
Postprandial Aminogenic Insulin and Glucagon Secretion can Stimulate Glucose Flux in Humans (2019) https://diabetesjournals.org/diabetes/article/68/5/939/39786/Postprandial-Aminogenic-Insulin-and-Glucagon Dietary protein does not cause raised glucose up to 4 hours post meal while glucagon was released more during that time
-
Glucagon acutely regulates hepatic amino acid catabolism https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7560169/ (2020) Our study demonstrates that lack of glucagon signaling as well as hepatic steatosis results in impaired amino acid metabolism, and, furthermore, that glucagon plays a role in the minute-to-minute regulation of amino acid turnover and urea formation, which is impaired in hepatic steatosis. Our results therefore support the existence of a liver-alpha cell axis [5,8] in which glucagon controls plasma levels of amino acids through activation of hepatic ureagenesis. Disruption of the axis, e.g., due to increased hepatic fat accumulation, results in hyperaminoacidemia and hyperglucagonemia, and given that the hyperglycemic effects of hyperglucagonemia seem to be preserved in this situation [46], these observations point toward a diabetogenic role of steatosis-induced disruption of the liver-alpha cell axis [35].
-
Glucagon Stimulates gluconeogenesis by InsPR3R-I mediated hepatic lipolysis (2020) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7101062/ While it is well-established that alterations in the portal vein insulin/glucagon ratio play a major role in causing dysregulated hepatic glucose metabolism in type 2 diabetes (T2D)1–3, the mechanisms by which glucagon alters hepatic glucose production and mitochondrial oxidation remain poorly understood. Here we show that glucagon stimulates hepatic gluconeogenesis by increasing hepatic adipose triglyceride lipase activity, intrahepatic lipolysis, hepatic acetyl-CoA content, and pyruvate carboxylase flux, while also increasing mitochondrial fat oxidation, mediated by stimulation of the inositol triphosphate receptor-1 (InsP3R-I).
-
Modeling of dysregulated glucagon secretion in T2D by considering mitochondrial alterations in pancreatic alpha-cells (2020) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7029933/ In addition to glucose and FFA studied here, amino acids play an important role in glucagon secretion. There is a large body of evidence that glucagon regulates amino acid metabolism at a systemic level, probably even more efficiently than the glucose homeostasis [69,70]. By way of a feedback loop, an elevation in circulating amino acids causes enormous glucagon secretion, known for decades [71], e.g. an intravenous arginine infusion of 5 g may result in a 10-fold increase in plasma glucagon level [72]. Amino acids also promote α-cell proliferation via a nutrient-sensing circuit [73,74]. Whereas the signalling role of amino acids on glucagon secretion is well established, its contribution to the energy production in α-cells is of much less importance. In particular, in hypoglycaemia, it is hardly to expect that amino acids would be used for ATP production in α-cells, especially in significant quantities.
-
Mitochondrial dysfunction in Pancreatic Alpha and Beta Cells Associated with T2D (2020) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7764865/ Therefore, amino acids add another layer of complexity to the islet cell metabolism and hormone secretion dynamics, and they represent a viable route for further investigation, including computational modeling approaches. Understanding the mechanisms by which amino acids along with other nutrients regulate islet hormone secretion has the potential to assess novel targets for future diabetes therapies [61].
-
Tracking the carbons supplying gluconeogenesis (2020) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7573258/ Circulating glycerol concentrations and glycerol turnover were consistently higher in subjects with T2DM compared with healthy controls in three studies that assessed the parameters (52, 53, 87). Insulin resistance leads to increased lipolysis, which allows for greater release of glycerol into circulation, and T2DM is routinely linked with increased fat mass (88). This would allow glycerol to be a net carbon contributor to gluconeogenesis in T2DM.
-
Postprandial Effects of a Whey Protein-Based Multi-Ingredient Nutritional Drink Compared with a normal breakfast on glucose, insulin, and active GLP-1 response among type 2 diabetic subjects: a crossover randomized control trial (2021) https://www.cambridge.org/core/journals/journal-of-nutritional-science/article/postprandial-effects-of-a-whey-proteinbased-multiingredient-nutritional-drink-compared-with-a-normal-breakfast-on-glucose-insulin-and-active-glp1-response-among-type-2-diabetic-subjects-a-crossover-randomised-controlled-trial/8059903DC077C7646B0EBC154BFCD1C3 The present findings indicated that WD provided a better postprandial glvcaemic response and active GLP-1 levels without elevating the insulinaemic excursions compared with ordinary breakfast in patients with T2DM. These could be implied that the nutrition formula containing a mixture of slowly digested carbohydrates and whey protein might be shown to exert beneficial glycaemic management when compared with normal breakfast when used as a meal replacement.
-
The-Liver-Alpha-Cell Axis after a mixed meal and during weight loss in T2D (2021) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8494406/ Glucagon receptors are almost exclusively found in the liver (and in the kidney) where glucagon increases endogenous glucose production and ureagenesis (23). In response to a glucagon receptor antagonist, plasma amino acid levels rise within hours (24). It has, therefore, been proposed that glucagon plays an important role in acute amino acid metabolism (i.e. ureagenesis) in the liver (23). This regulatory cycle is thought to be important for maintaining normal amino acid and ammonia levels during protein abundance for example, after protein-rich meals.
-
Modeling the Amino Acid Effect on Glucagon Secretion from Pancreatic Alpha Cells (2022) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9028923/ The model predictions successfully reproduce the experimental results of Zhang et al. [35], showing that AAs contribute substantially to the glucagon response of pancreatic alpha cells. An increase in AA levels resulted in a robust maximal glucagon response up to 7-fold stronger than the maximal glucagon response elicited by glucose alone. The mechanisms responsible for this phenomenon are modeled by glutamate-induced modulation of MP oscillations (via iGluR-induced depolarization) and autocrine glucagon-induced stimulation of GcgR-bound tmACs, which increases intracellular cAMP concentration. Model predictions showed that AAs enhance Ca2+ entry by synergistically increasing the frequency and amplitude of MP oscillations. However, modulation of MP mainly indirectly affects glucagon exocytosis, whereas most of the net effect is due to increased cAMP concentration.