This arose from a discussion with Peter@hyperlipid. I wondered what you make of it. It’s a wild idea, but with some science behind it. It references protons but you don’t have to know the theory. In fact, it’s pretty much an alternative theory why n-6 PUFAs make us fat on a high-carb diet, independent from protons.
Jason Fung points out that the assumption is weird that muscle cells become insulin resistant while other cells (like the ones in the liver producing triglycerides out of glucose, or lipid cells storing fat and not releasing it) remain apparently perfectly insulin sensitive, and I agree. He thinks that the cells are simply full of glucose, where I disagree. (After all, a few weeks of keto do not restore insulin sensitivity, while it certainly depletes glycogen stores.) But he still has a point that something doesn’t check out here.
My personal issue with the protons theory is that the RET happens in ATP production in the mitochondria, which is round the clock while food intake is time restricted. Most food has to be stored (glucose as glycogen or fat in chylomicrons) for some time. Now why are saturated fats immediately more satiating (no pun intended) as evidenced by protons, the croissant diet and https://doi.org/10.1038/nutd.2016.1 in a meal if the effects from the mitochondria should be spread out over many hours? Why would fatty acids with high F:N ratio (as per Peters protons theory) produce local insulin resistance at fat cells (or possibly liver cells, preventing triglyceride synthesis) but not affect muscle cells, causing slower glucose uptake?
We’re missing something here. And Fungs brilliant idea that the all cells are insulin sensitive but muscle cells simply don’t accept glucose for some other reason explains a LOT, if we find a solid explaination why the cells won’t accept glucose. And here it is:We replace Fungs assumption that the cells are crammed with glucose with the assumption that glycogenesis is hampered. (Lack of glycogenesis has been conjectured to be the fundamental mechanism of insulin resistance, https://www.ncbi.nlm.nih.gov/pubmed/18220643).
Obviously we have an intra-cell homeostasis that will convert extra glucose to glycogen, and glycogen to glucose if needed. Assume that we have a thrown a wrench in the spokes here, and only very high insulin and/or significantly higher glucose concentration will force glycogenesis. The effects would be exactly the ones we see in (pre)diabetics:
- Blood glucose rises, insulin rises. Eventually the high insulin levels will activate glycogenesis anyway, but until then the liver has converted a lot of the glucose to triglycerides already (because that pathway is not blocked). Sounds familiar? Most glucose goes to fat and is not available as energy.
- Insulin is remains high after glucose clearance, glycogen stores are almost empty, muscles need glucose (insulin blocks ketone production), we go hypo and need to eat again.
- Neoglucogenesis is probably running overtime but can’t satisfy the demand, because our body is not designed to run on glucose manufactured from protein with no ketones to help out. (That would explain why neoglucogenesis runs in overdrive for many diabetics.)
- This all fits. Glucose and triglycerides do their damage in the pancreas, and eventually we’ll have diabetes.
- For diabetics, insulin shots jumpstart glycogenesis and relieve the symptoms temporarily.
- It might explain the dawn effect, where the liver pumps some extra glycogen into the bloodstream (if I understand it correctly). If glycogen stores in the cells are low, we would see a spike of glucose if the liver attempts to jumpstart our muscle cells.
Now, why would glycogenesis specifically be hampered while other cells remain insulin sensitive? Enter n-6 PUFAs. Calcium (cAMP) blocks glycogenesis, and both linoleic acid and arachidonic acid mess with calcium in our body (https://www.ncbi.nlm.nih.gov/pubmed/18718873 - I admit I’m a bit fuzzy on this one). (But you’ve got to wonder if it affects artherosclerosis and calcification.)
Plus arachidonic acid activates our immune system and increases oxidative stress (https://iubmb.onlinelibrary.wiley.com/doi/full/10.1002/iub.1428) which could mess with ROS signalling. Could this be the reason why n-6 PUFAs block glycogenesis, while oleic acid and saturated fats don’t have this effect?
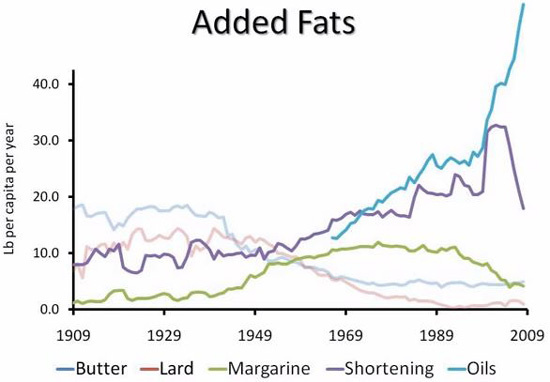
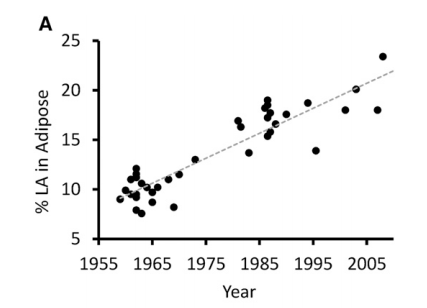
PUFAs will accumulate in our bodies with time in our fat tissue as per https://www.ncbi.nlm.nih.gov/pubmed/26567191, and it gets used as building blocks for cell membranes. So our body becomes a big PUFA store with time, boom! glucose metabolism is kaputt.
Thoughts?