My question is ‘what might limit ketogenesis in long-term fat adapted individuals?’ A secondary related question is it something to be concerned about? Do we just go with the flow and accept it as a natural outcome? KCKO, etc.
The experience of greatly reduced ketones after being on keto for several years and presumably fat adapted has been noted by multiple posters here on the forum.
This is going to be a long post. So I shall break it up into pieces. This is the first piece.
Let’s first get the initial parameters out of the way. I’m talking about folks who are in long-term ketosis. These people have eaten a standard keto diet defined as ‘high fat’, ‘moderate protein’ and ‘very low/no carbs’. Pretty much Phinney and Volek’s definition of the nutrional keto diet. I’m also talking about folks who have remained consistently in nutritional ketosis for several consecutive years. By ‘consistently’ I mean specifically keeping carb intake and resultant glucose/insulin levels sufficiently low as not to interfere with continuous ketosis. This excludes anyone doing alternative keto diets such as: TKD, CKD and HPKD. Or any other iterations that intentionally/accidentally interrupt continuous ketosis whether on a regular basis or not. It’s your business not mine if you’re doing any of that, but I don’t think your experiences are relevant to answering my questions.
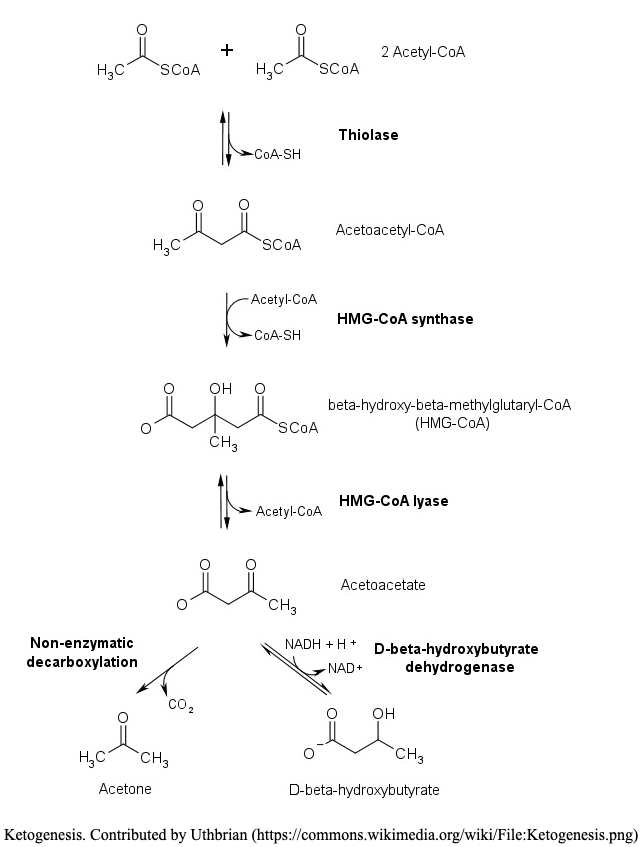
From Biochemistry, Ketogenesis
Mechanism
Ketogenesis occurs primarily in the mitochondria of liver cells. Fatty acids are brought into the mitochondria via carnitine palmitoyltransferase (CPT-1) and then broken down into acetyl CoA via beta-oxidation. Two acetyl-CoA molecules are converted into acetoacetyl-CoA via the enzyme thiolase; this is also known as acetyl coenzyme A acetyltransferase (ACAT). Afterward, acetoacetyl-CoA is converted to HMG-CoA via the enzyme HMG-CoA synthase. HMG-CoA lyase then converts HMG-CoA to acetoacetate. Acetoacetate can be converted to either acetone through non-enzymatic decarboxylation or to beta-hydroxybutyrate via beta-hydroxybutyrate dehydrogenase.
Acetoacetate and beta-hydroxybutyrate are the two ketone bodies used by the body for energy. Once they reach extrahepatic tissues, beta-hydroxybutyrate is converted to acetoacetate via the enzyme beta-hydroxybutyrate dehydrogenase, and acetoacetate is converted back to acetyl-CoA via the enzyme beta-ketoacyl-CoA transferase. Acetyl-CoA goes through the citric acid cycle, and after oxidative phosphorylation, produces 22 ATP per molecule. Acetone does not convert back to acetyl-CoA, so it is either excreted through urine or exhaled.
Right away, I can see multiple points of potential issues. Fatty acids are delivered to the mitochondria for processing via Carnitine Palmitoyltransferase (CPT-1). What happens if CPT-1 is not functioning normally or not produced in sufficient quantity? What if CPT-1 is ‘decoupled’ and turning fat into lost heat, as described by Bikman in his talks about BAT? What if Bikman is substantially correct that being in ketosis gives one a ‘metabolic advantage’ and that advantage is the decoupling of CPT-1 such that you’re blowing off more energy in the form of wasted heat rather than ketone synthesis? That could explain the whole thing in a single sentence. Maybe.
According to Biochemistry, Ketogenesis, after being brought into the mitochondria via CPT-1 fatty acids are then 'broken down into acetyl CoA via beta-oxidation. But that may be an outmoded description because according to The Definitive Guide:
Acetyl-CoA Formation through Fatty Acids
Acetyl-CoA formation is also said to occur via fatty acid catabolism; however, it is now understood that this acetyl-CoA is a product of carbohydrate metabolism. As acetyl-CoA can be converted into lipids and vice versa it is sometimes confused with a separate role; its true role is as a monosaccharide (glucose) metabolism catalyst.
This agrees with a comment I read from @amber in one of her blog posts about why we might have low ketones years into ketosis - the limits of acetyl-CoA due to our reduced carb consumption. Sorry I can’t cite the post, but I’ve noted it in another post, so may be able to find it and add it here.
So if we can expect lower acetyl-CoA as a result of low/no carb intake, how exactly does that impact the very first step of the process? Is some fat converted to acetyl-CoA? If so, how much? Does that amount vary individually? Or is no fat converted and gluconeogenesis has to make some glucose so it can catabolize into acetyl-CoA so the fat can catabolize further? Do we, in fact, have to synthesize some amount of glucose in order to synthesize some amount of acetyl-CoA to burn fat?
We’re not even down to:
- Two acetyl-CoA molecules are converted into acetoacetyl-CoA via the enzyme thiolase. What if you have a thiolase deficiency?
- Acetoacetyl-CoA is converted to HMG-CoA via the enzyme HMG-CoA synthase. What if you have a HMG-CoA synthase deficiency?
- HMG-CoA lyase then converts HMG-CoA to acetoacetate. What if…
I can see this is going down a rabbit hole real fast. More to come. 



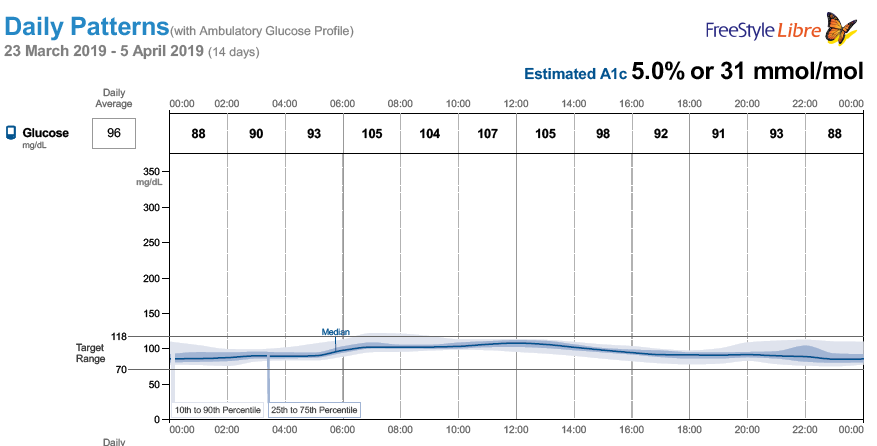
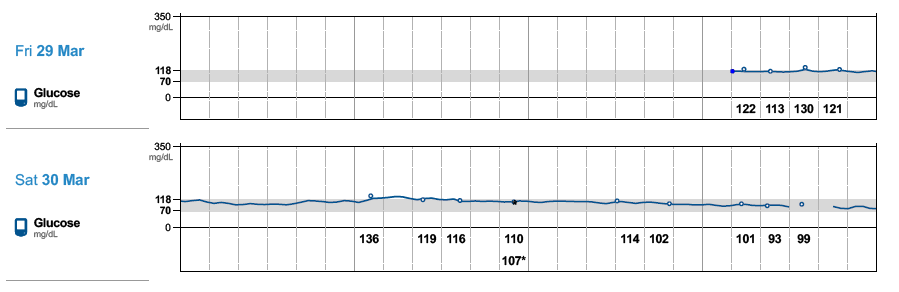
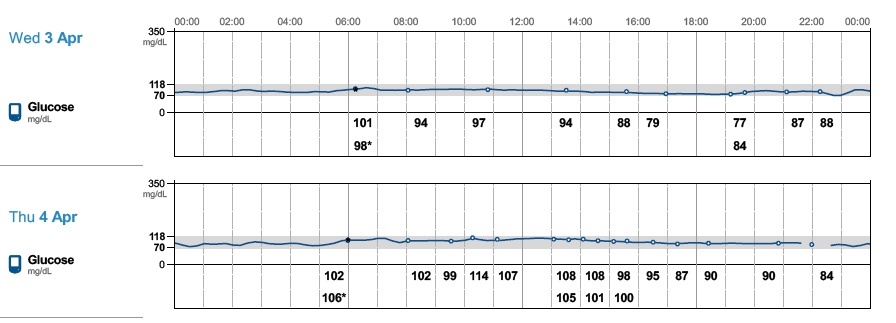
 } and see if there are any positive changes. I am starting to believe GNG is very real in my case. Every expert out there says GNG is not something be concerned about it. Another alternative will be to try low dose metformin to reduce liver dumping sugars but its by prescription-only in US. The direction my A1C is headed, I might be able to get a prescription, lol! I just don’t like the idea of experimenting with my body but doing things for a week to test might be ok, I guess. Just laying out as much info as possible hoping others can share their view and experience with this situation.
} and see if there are any positive changes. I am starting to believe GNG is very real in my case. Every expert out there says GNG is not something be concerned about it. Another alternative will be to try low dose metformin to reduce liver dumping sugars but its by prescription-only in US. The direction my A1C is headed, I might be able to get a prescription, lol! I just don’t like the idea of experimenting with my body but doing things for a week to test might be ok, I guess. Just laying out as much info as possible hoping others can share their view and experience with this situation.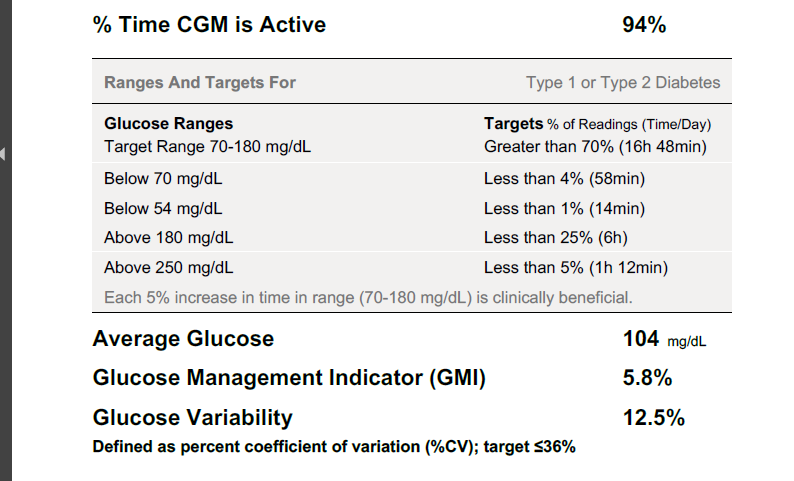
{kind=link}